SearchUser loginOffice of CitizenRest in Peace,
Who's new
Subscribe |
Dear President Obama: Reading Material For Your Flight Home From Cancer-causing ClevelandSubmitted by Norm Roulet on Tue, 02/22/2011 - 09:08.
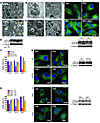
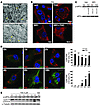
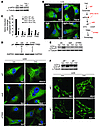
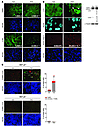
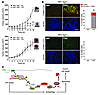
Autophagy can promote cell survival or cell death, but the molecular basis underlying its dual role in cancer remains obscure. Here we demonstrate that Δ9-tetrahydrocannabinol (THC), the main active component of marijuana, induces human glioma cell death through stimulation of autophagy. Our data indicate that THC induced ceramide accumulation and eukaryotic translation initiation factor 2α (eIF2α) phosphorylation and thereby activated an ER stress response that promoted autophagy via tribbles homolog 3–dependent (TRB3-dependent) inhibition of the Akt/mammalian target of rapamycin complex 1 (mTORC1) axis. We also showed that autophagy is upstream of apoptosis in cannabinoid-induced human and mouse cancer cell death and that activation of this pathway was necessary for the antitumor action of cannabinoids in vivo. These findings describe a mechanism by which THC can promote the autophagic death of human and mouse cancer cells and provide evidence that cannabinoid administration may be an effective therapeutic strategy for targeting human cancers. IntroductionMacro-autophagy, hereafter referred to as “autophagy,” is a highly conserved cellular process in which cytoplasmic materials — including organelles — are sequestered into double-membrane vesicles called autophagosomes and delivered to lysosomes for degradation or recycling (1). In many cellular settings, triggering of autophagy relies on the inhibition of mammalian target of rapamycin complex 1 (mTORC1), an event that promotes the activation (de-inhibition) of several autophagy proteins (Atgs) involved in the initial phase of membrane isolation (1). Enlargement of this complex to form the autophagosome requires the participation of 2 ubiquitin-like conjugation systems. One involves the conjugation of ATG12 to ATG5 and the other of phosphatidylethanolamine to LC3/ATG8 (1). The final outcome of the activation of the autophagy program is highly dependent on the cellular context and the strength and duration of the stress-inducing signals (2–5). Thus, besides its role in cellular homeostasis, autophagy can be a form of programmed cell death, designated “type II programmed cell death,” or play a cytoprotective role, for example in situations of nutrient starvation (6). Accordingly, autophagy has been proposed to play an important role in both tumor progression and promotion of cancer cell death (2–4), although the molecular mechanisms responsible for this dual action of autophagy in cancer have not been elucidated. Δ9-Tetrahydrocannabinol (THC), the main active component of marijuana (7), exerts a wide variety of biological effects by mimicking endogenous substances — the endocannabinoids — that bind to and activate specific cannabinoid receptors (8). One of the most exciting areas of research in the cannabinoid field is the study of the potential application of cannabinoids as antitumoral agents (9). Cannabinoid administration has been found to curb the growth of several types of tumor xenografts in rats and mice (9, 10). Based on this preclinical evidence, a pilot clinical trial has been recently run to investigate the antitumoral action of THC on recurrent gliomas (11). Recent findings have also shown that the pro-apoptotic and tumor growth–inhibiting activity of cannabinoids relies on the upregulation of the transcriptional co-activator p8 (12) and its target the pseudo-kinase tribbles homolog 3 (TRB3) (13). However, the mechanisms that promote the activation of this signaling route as well as the targets downstream of TRB3 that mediate its tumor cell–killing action remain elusive. In this study we found that ER stress–evoked upregulation of the p8/TRB3 pathway induced autophagy via inhibition of the Akt/mTORC1 axis and that activation of autophagy promoted the apoptotic death of tumor cells. The uncovering of this pathway, which we believe is novel, for promoting tumor cell death may have therapeutic implications in the treatment of cancer. ResultsAutophagy mediates THC-induced cancer cell death. As a first approach to gain insight into the morphological changes induced in cancer cells by cannabinoid administration, we performed electron microscopy analysis of U87MG human astrocytoma cells. Interestingly, double membrane vacuolar structures with the morphological features of autophagosomes were observed in THC-treated cells (Figure 1, A–C). The conversion of the soluble form of LC3 (LC3-I) to the lipidated and autophagosome-associated form (LC3-II) is considered one of the hallmarks of autophagy (1), and thus we observed the occurrence of LC3-positive dots as well as the appearance of LC3-II (Figure 1D) in cannabinoid-challenged cells. In addition, co-incubation with the lysosomal protease inhibitors E64d and pepstatin A, which blocks the last steps of autophagic degradation (14), enhanced THC-induced accumulation of LC3-II (Figure 1E), confirming that cannabinoids induce dynamic autophagy in U87MG cells. Furthermore, incubation with the cannabinoid receptor 1 (CB1) antagonist SR141716 prevented THC-induced LC3 lipidation and formation of LC3 dots (Figure 1D), indicating that induction of autophagy by cannabinoids relies on CB1 receptor activation. Inhibition of autophagy prevents THC-induced cancer cell death. (A–C) Effect of THC on U87MG cell morphology. Representative electron microscopy photomicrographs are shown (6 h). Scale bars: 500 nm. Note the presence of early (A, open arrows, and B) and late (A, filled arrows, and C) autophagosomes in THC-treated but not vehicle-treated (veh-treated) cells. (D) Top: Effect of SR141716 (SR1; 1 μM) and THC on LC3 immunostaining (green) in U87MG cells (18 h; n = 3). The percentage of cells with LC3 dots relative to the total cell number is shown in the corner of each panel (mean ± SD). Scale bar: 20 μm. Bottom: Effect of SR1 and THC on LC3 lipidation in U87MG cells (18 h; n = 3). (E) Effect of E64d (10 μM) and pepstatin A (PA; 10 μg/ml) on THC-induced LC3 lipidation in U87MG cells (18 h; n = 3). (F and G) Effect of THC treatment and transfection with control siRNAs (siC) or ATG1-selective siRNAs (siATG1) on cell viability (F; mean ± SD; n = 3), LC3 immunostaining (G, left panels; 18 h; percentage of cells with LC3 dots relative to the total number of cells cotransfected with a red fluorescent control siRNA, mean ± SD; n = 3; scale bar: 20 μm), and LC3 lipidation (G, right panel; 18 h; n = 3) in U87MG cells. (H and I) Effect of THC on cell viability (H; mean ± SD; n = 3), LC3 immunostaining (I, left panels; 18 h; percentage of cells with LC3 dots relative to the total cell number, mean ± SD; n = 3; scale bar: 20 μm), and LC3 lipidation (I, right panel; 18 h; n = 3) in Atg5+/+ and Atg5–/– RasV12/T-large antigen MEFs. *P < 0.05 and **P < 0.01 compared with THC-treated U87MG (D) and Atg5+/+ (H and I) cells and compared with siC-transfected, THC-treated U87MG cells (F and G). THC concentration was 6 μM. Since autophagy has been implicated in promotion and inhibition of cell survival, we next investigated its participation in the cancer cell death–inducing action of THC. Pharmacological inhibition of autophagy at different levels (Supplemental Figure 1, A–C; supplemental material available online with this article; doi:10.1172/JCI37948DS1) or selective knockdown of ATG1 (an essential protein in the initiation of autophagy; ref. 1) (Figure 1, F and G), ATG5 (an essential protein in the formation of the autophagosome; ref. 1) (Supplemental Figure 1, D–F), or AMBRA1 (a recently identified beclin-1–interacting protein that regulates autophagy; ref. 15) (Supplemental Figure 1, D–F) strongly reduced cannabinoid-induced autophagy and cell death. Moreover, transformed Atg5-deficient mouse embryonic fibroblasts (MEFs), which are defective in autophagy (16), were more resistant than their wild-type counterparts to THC-induced cell death (Figure 1H) and did not undergo autophagy upon cannabinoid treatment (Figure 1I). Taken together, these findings demonstrate that autophagy plays a prominent role in THC-induced cancer cell death. THC induces autophagy via ER stress–dependent upregulation of p8 and TRB3. In addition to the presence of autophagosomes, electron microscopy analysis of cannabinoid-treated cells revealed the presence of numerous cells with dilated ER (Figure 2A). In line with this observation, immunostaining of the ER luminal marker protein disulphide isomerase (PDI) showed a striking dilation in the ER of THC-treated U87MG cells (Figure 2B), an event that was associated with an increased phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α), a hallmark of the ER stress response (17) (Figure 2C). In addition, THC-induced ER dilation and eIF2α phosphorylation were prevented by pharmacological blockade of the CB1 receptor (Figure 2, B and C). ER stress precedes autophagy in cannabinoid action. (A) Effect of THC on U87MG cell morphology. Note the presence of the dilated ER in THC- but not vehicle-treated cells (6 h). Arrows point to the ER. Scale bars: 500 nm. (B) Effect of SR1 (1 μM) and THC on PDI immunostaining (red) in U87MG cells (8 h; n = 3). The percentage of cells with PDI dots relative to the total cell number is shown in the corner of each panel (mean ± SD). Scale bar: 20 μm. (C) Effect of SR1 (1 μM) on THC-induced eIF2α phosphorylation of U87MG cells (3 h; OD relative to vehicle-treated cells, mean ± SD; n = 3). (D) Effect of THC on PDI (red) and LC3 (green) immunostaining in U87MG cells (n = 3). The percentage of cells with PDI or LC3 dots relative to total cell number at each time point (mean ± SD) is shown. Scale bar: 20 μm. (E) Effect of THC on eIF2α phosphorylation and LC3 lipidation in U87MG cells (n = 3). **P < 0.01 compared with THC-treated (B) or vehicle-treated (C and D) cells. Time-course analysis of PDI and LC3 immunostaining, eIF2α phosphorylation, and LC3 lipidation of cannabinoid-treated cells revealed that ER stress occurred earlier than autophagy (Figure 2, D and E). Of interest, cannabinoid administration produced similar activation of ER stress and autophagy, as well as cell death, in other human astrocytoma cell lines (Supplemental Figure 2, A–F), a primary culture of human glioma cells (Supplemental Figure 2, G–I), and several human cancer cell lines of different origin, including pancreatic cancer (Supplemental Figure 2, J–L), breast cancer, and hepatoma (data not shown). However, neither ER dilation nor eIF2α phosphorylation or autophagy was evident in normal, nontransformed primary astrocytes (Supplemental Figure 3), which are resistant to cannabinoid-induced cell death (13). We next investigated whether activation of ER stress is involved in the induction of autophagy in response to cannabinoid treatment of cancer cells. We have previously shown that THC-induced accumulation of de novo–synthesized ceramide, an event that occurs in the ER (18), leads to upregulation of the stress-regulated protein p8 and its ER stress–related downstream targets, ATF4, CHOP, and TRB3, to induce cancer cell death (13). Of importance, incubation with ISP-1 (a selective inhibitor of serine palmitoyltransferase, the enzyme that catalyzes the first step of sphingolipid biosynthesis; ref. 18) prevented ceramide accumulation (Supplemental Figure 4A); THC-induced ER dilation (Supplemental Figure 4B); eIF2α phosphorylation (Figure 3A); p8, ATF4, CHOP, and TRB3 upregulation (Supplemental Figure 4C); and autophagy (Figure 3B), supporting that ceramide accumulation is involved in cannabinoid-triggered ER stress and autophagy. We also verified by means of RNA interference that CaCMKKβ — which had been previously implicated in activating autophagy in response to ER stress–associated calcium release (19) — was not involved in THC-induced autophagy and cell death (data not shown). As phosphorylation of eIF2α on Ser51 attenuates general protein synthesis while enhancing the expression of several ER stress response genes (17), we used cells derived from eIF2α S51A knockin mice to test whether eIF2α phosphorylation regulates the expression of p8 and its downstream targets. In agreement with this hypothesis, THC treatment (which promoted ceramide accumulation in both wild-type and eIF2α S51A immortalized MEFs; Supplemental Figure 5A) triggered p8, ATF4, CHOP, and TRB3 upregulation (Figure 3C) as well as autophagy (Supplemental Figure 5B) in wild-type cells but not in their eIF2α S51A counterparts. THC induces autophagy via ER stress–evoked p8 and TRB3 upregulation. (A and B) Effect of ISP-1 (1 μM) on THC-induced eIF2α phosphorylation (A; 3 h; n = 3) and LC3 immunostaining (B, left panels; 18 h; percentage of cells with LC3 dots relative to the total cell number, mean ± SD; n = 3; scale bar: 20 μm) in U87MG cells. sip8, p8-selective siRNA; siTRB3, TRB3-selective siRNA. (C) Effect of THC on p8, ATF4, CHOP, and TRB3 mRNA levels of eIF2α WT and eIF2α S51A MEFs as determined by real-time quantitative PCR (8 h; n = 3). Numbers indicate the mean fold increase ± SD relative to vehicle-treated eIF2α WT MEFs. (D) Top: Analysis of p8 and TRB3 mRNA levels. Results from a representative RT-PCR experiment are shown. The numbers indicate gene expression levels as determined by real-time quantitative PCR (mean fold change ± SD relative to siC-transfected cells; n = 5). Bottom: Effect of THC on LC3 immunostaining (green) of U87MG cells transfected with siC, sip8, or siTRB3 (18 h; n = 4). The percentage of cells with LC3 dots relative to cells cotransfected with a red fluorescent control siRNA is shown in each panel (mean ± SD). Scale bar: 20 μm. (E) Effect of THC on LC3 lipidation in U87MG cells transfected with siC, sip8, or siTRB3 (18 h; n = 6). (F) Effect of THC on LC3 lipidation (top; 18 h; n = 5) and LC3 immunostaining (bottom; 18 h; percentage of cells with LC3 dots relative to the total cell number, mean ± SD; n = 4; scale bar: 40 μm) in p8+/+ or p8–/– MEFs. *P < 0.05 and **P < 0.01 compared with THC-treated U87MG (B), eIF2α WT (C), or p8+/+ (F) cells and compared with siC-transfected, THC-treated U87MG cells (D). We subsequently asked whether p8 and its downstream targets regulate autophagy. Knockdown of p8 or TRB3 prevented THC-induced autophagy (Figure 3, D and E) but not ER dilation (Supplemental Figure 4D) in U87MG cells. Furthermore, THC induced autophagy in p8+/+ but not p8-deficient transformed MEFs (Figure 3F and Supplemental Figure 5C). Altogether, these findings reveal that THC induces autophagy of cancer cells via activation of an ER stress–triggered signaling route that involves stimulation of ceramide synthesis de novo, eIF2α phosphorylation, and p8 and TRB3 upregulation. THC inhibits Akt and mTORC1 via TRB3. Inhibition of mTORC1 is considered a key step in the early triggering of autophagy (6). We therefore tested whether cannabinoid-induced upregulation of the p8 pathway leads to autophagy via inhibition of this complex. THC treatment of U87MG cells reduced the phosphorylation of p70S6 kinase (a well-established mTORC1 substrate) and the ribosomal protein S6 (a well-established p70S6 kinase substrate) (Figure 4, A and C), indicating that mTORC1 is inhibited in cannabinoid-challenged cells. In addition, the cannabinoid-induced decrease in p70S6 kinase and S6 phosphorylation, autophagy, and cell death were not evident in Tsc2–/– cells, in which mTORC1 is constitutively active (20) (Figure 4B and Supplemental Figure 6, A and B), further supporting a major role for mTORC1 inhibition in the induction of autophagic cell death by cannabinoids. THC inhibits the Akt/mTORC1 pathway via TRB3. (A) Effect of THC on p70S6K and S6 phosphorylation of U87MG cells (n = 6). (B) Effect of THC on cell viability (left panel; 24 h; mean ± SD; n = 6) and LC3 lipidation (right panel; 18 h; n = 4) in Tsc2+/+ and Tsc2–/– MEFs. (C) Effect of THC on Akt, TSC2, PRAS40, p70S6K, and S6 phosphorylation of U87MG cells (18 h; OD relative to vehicle-treated cells, mean ± SD; n = 7). (D) Effect of THC on cell viability (left panel; 24 h; mean ± SD; n = 4) and LC3 lipidation (right panel; 18 h; n = 4) of pBABE and myristoylated Akt (myr-Akt) MEFs. (E) Effect of THC on Akt co-immunoprecipitation with TRB3 in U87MG cell extracts (8 h; OD relative to vehicle-treated cells, mean ± SD; n = 9; input: TRB3). (F and G) Effect of THC on Akt, TSC2, PRAS40, p70S6K, and S6 phosphorylation and LC3 lipidation (G only) of siC- and siTRB3-transfected (F; 18 h; OD relative to vehicle-treated siC-transfected U87MG cells, mean ± SD; n = 7; upper panel shows an analysis of TRB3 mRNA levels) and EGFP (Ad-EGFP) or rat TRB3 (Ad-TRB3) adenoviral vector–infected (G; 18 h; OD relative to vehicle-treated Ad-EGFP–infected U87MG cells, mean ± SD; n = 4; upper panel shows an analysis of rTRB3 mRNA levels) U87MG cells. (H) Effect of THC on Akt, p70S6K, and S6 phosphorylation of p8+/+ and p8–/– MEFs (n = 7). *P < 0.05 and **P < 0.01 compared with THC-treated Tsc2+/+ (B) and pBABE (D) MEFs and compared with vehicle-treated (C and E), vehicle-treated siC-transfected (F), or Ad-EGFP–infected (G) U87MG cells. The protein kinase Akt positively regulates the activity of the mTORC1 complex by phosphorylating and inhibiting TSC2 and PRAS40 (a well-established Akt substrate within the mTORC1 complex). Thus, Akt inhibition decreases mTORC1 activity and promotes autophagy (20). In line with this idea, THC decreased the phosphorylation of Akt, TSC2, and PRAS40 as well as p70S6 kinase and S6 (Figure 4C). This inhibition of the Akt/mTORC1 pathway was abrogated by incubation with a CB1 receptor antagonist (Supplemental Figure 6C) or a ceramide synthesis inhibitor (Supplemental Figure 6D). Likewise, cells overexpressing a myristoylated (constitutively active) form of Akt were resistant to THC-induced mTORC1 inhibition, autophagy, and cell death (Figure 4D and Supplemental Figure 6, E and F), further supporting that THC induces autophagy via Akt inhibition. Since TRB3 has been shown to directly interact with and inhibit Akt (21, 22), we investigated whether upregulation of TRB3 was responsible for THC-induced Akt/mTORC1 inhibition. Several observations support that this is indeed the case: (a) THC increased the amount of Akt coimmunoprecipitated with TRB3 from U87MG extracts (Figure 4E), (b) knockdown of TRB3 prevented the effect of THC on Akt, TSC2, PRAS-40, p70S6 kinase, and S6 phosphorylation (Figure 4F), and (c) TRB3 overexpression decreased Akt, TSC2, PRAS40, p70S6 kinase, and S6 phosphorylation, enhanced the inhibitory effect of THC on the phosphorylation of these proteins, and promoted autophagy (Figure 4G). In line with these observations, THC failed to inhibit Akt, p70S6 kinase, and S6 phosphorylation of eIF2α S51A knockin or p8-deficient MEFs, in which TRB3 did not become upregulated upon cannabinoid treatment (Figure 4H and Supplemental Figure 6, G and H). Altogether, these data demonstrate that upregulation of p8 and TRB3 induce autophagy of tumor cells via inhibition of the Akt/mTORC1 pathway. THC-induced autophagy promotes the apoptotic death of cancer cells. While analyzing the mechanism of cannabinoid cell-killing action, we observed that incubation with the pan-caspase inhibitor ZVAD-fmk prevented cell death to the same extent as genetic (Figure 5A) or pharmacological (Supplemental Figure 7) inhibition of autophagy. Furthermore, Bax/Bak double knockout (DKO) immortalized MEFs, which are protected against mitochondrial apoptosis (23), were resistant to THC-induced cell death and apoptosis (Figure 5B) but underwent eIF2α phosphorylation and autophagy (Figure 5C) upon THC treatment. We therefore investigated whether cannabinoid-induced autophagy promoted the apoptotic death of cancer cells. Time-course analysis of LC3 and active caspase-3 immunostaining in U87MG cells revealed that autophagy preceded the appearance of apoptotic features in THC-treated cells (Figure 5D). In addition, selective knockdown of ATG1 (Figure 5D) as well as of AMBRA1 or ATG5 (Supplemental Figure 8) prevented THC-induced caspase-3 activation. Moreover, unlike their wild-type counterparts, Atg5-deficient immortalized MEFs did not undergo phosphatidylserine translocation to the outer leaflet of the plasma membrane (Figure 5E), loss of mitochondrial membrane potential (Figure 5F), or increased production of reactive oxygen species (Supplemental Figure 9) in response to cannabinoid treatment. These findings indicate that activation of the autophagy-mediated cell death pathway occurs upstream of apoptosis in cannabinoid antitumoral action. Autophagy is upstream of apoptosis in cannabinoid-induced cancer cell death. (A) Effect of THC and the pan-caspase inhibitor ZVAD (10 μM) on the viability of Atg5+/+ and Atg5–/– MEFs (36 h; percentage of viable cells relative to the corresponding Atg5+/+ vehicle-treated cells, mean ± SD; n = 3). (B) Effect of THC on the apoptosis of Bax/Bak WT and Bax/Bak DKO MEFs as determined by cytofluorometric analysis of Annexin V/propidium iodide (PI) (24 h; mean ± SD; n = 3). The mean ± SD percentage of Annexin V–positive/PI-positive and Annexin V–positive, PI-negative cells is shown in the upper and lower corners, respectively. (C) Effect of THC on eIF2α phosphorylation (3 h; n = 3) and LC3 lipidation (18 h; n = 4) of Bax/Bak WT and DKO MEFs. (D) Left: Effect of THC on autophagy and apoptosis of U87MG cells transfected with siC or siATG1. Green bars, cells with LC3 dots; red bars, active caspase-3–positive cells; white bars, cells with both LC3 dots and active caspase-3 staining. Data correspond to the percentage of cells with LC3 dots (green bars), active caspase-3–positive cells (red bars), and cells with LC3 dots and active caspse-3 staining (white bars) relative to the total number of transfected cells at each time point (mean ± SD; n = 3). Right: Representative photomicrographs (36 h; scale bar: 20 μm). (E and F) Effect of THC on apoptosis (E; 24 h; n = 3) and loss of mitochondrial membrane potential as determined by DiOC6(3) staining (F; 24 h; n = 4) of Atg5+/+ and Atg5–/– MEFs. In E, the mean ± SD percentage of Annexin V–positive/PI-positive and Annexin V–positive, PI-negative cells is shown in the upper and lower corners, respectively. **P < 0.01 compared with THC-treated Atg5+/+ (A, E, and F) and Bax/Bak WT (B) MEFs and from THC-treated, siC-transfected cells (D). Activation of autophagy is necessary for cannabinoid antitumoral action in vivo. To determine the in vivo relevance of our findings, we first investigated whether THC promotes the activation of the above-described autophagy-mediated cell death pathway in U87MG cell–derived tumor xenografts, in which we have recently shown that cannabinoid treatment reduces tumor growth (specifically, THC administration for 14 days decreased tumor growth by 50%; ref. 13). Analysis of these tumors revealed that cannabinoid administration increases TRB3 expression and decreases S6 phosphorylation (Figure 6A). Likewise, formation of LC3 dots as well as increase in LC3-II and active caspase-3 immunostaining were observed in THC-treated, but not vehicle-treated, tumors (Figure 6B). THC activates the autophagic cell death pathway in vivo. (A) Effect of peritumoral THC administration on TRB3 and p-S6 immunostaining in U87MG tumors. TRB3- or p-S6–stained area normalized to the total number of nuclei in each section; numbers indicate the mean fold change ± SD; 18 sections were counted for each of 3 dissected tumors for each condition. Scale bar: 50 μm. (B) Left: Effect of peritumoral THC administration on LC3 and active caspase-3 immunostaining in U87MG tumors. Arrows point to cells with LC3 dots. The numbers indicate the percentage of active caspase-3–positive cells relative to the total number of nuclei in each section ± SD. Ten sections were counted for each of 3 dissected tumors for each condition. Scale bars: 20 μm. Right: Effect of peritumoral THC administration on LC3 lipidation in U87MG tumors. Representative samples from 1 vehicle-treated and 1 THC-treated tumor are shown. Numbers indicate the LC3-I and LC3-II OD values relative to vehicle-treated tumors (mean ± SD). n = 3. (C) Left: Effect of THC administration on LC3 immunostaining (green) and TUNEL (red) in RasV12/E1A p8+/+ and p8–/– tumor xenografts. Arrows point to cells with LC3 dots and TUNEL-positive nuclei. Right: Bar graph shows the percentage of TUNEL-positive nuclei or cells with TUNEL-positive nuclei and LC3 dots relative to the total number of nuclei in each section (mean ± SD). Eighteen sections were counted from 3 dissected tumors for each condition. Scale bars: 50 μm. Inset shows the magnification of 1 selected cell (arrows point to LC3 dots; scale bar: 10 μm). *P < 0.05 and **P < 0.01 compared with vehicle-treated tumors. To further investigate whether activation of the p8 pathway mediates cannabinoid antitumoral action, we also analyzed tumors derived from p8+/+ and p8–/– RasV12/E1A-transformed MEFs (in this case, THC administration for 8 days decreased by 45% the growth of p8+/+ tumors but had no significant effect on p8–/– tumors; ref. 13). THC treatment increased TRB3 expression, decreased S6 phosphorylation, and increased autophagy as well as TUNEL and active caspase-3 immunostaining in p8+/+ but not p8–/– tumors (Figure 6C and Supplemental Figure 10). Moreover, THC treatment enhanced the number of cells with LC3 dots and TUNEL-positive nuclei in p8+/+ but not in p8–/– tumors (Figure 6C). In order to verify the importance of autophagy for cannabinoid antitumoral action, we next generated tumors with Atg5+/+ and Atg5–/– RasV12/T-large antigen transformed MEFs. THC administration reduced by more than 80% the growth of tumors derived from wild-type cells but had no significant effect on those tumors generated by autophagy-deficient cells (Figure 7A). Furthermore, cannabinoid administration increased autophagy, TUNEL (Figure 7B), and active caspase-3 immunostaining (Supplemental Figure 11) in Atg5+/+ but not Atg5–/– tumors. Likewise, cannabinoid administration increased the number of cells with LC3 dots and TUNEL-positive nuclei in Atg5+/+ but not Atg5–/– tumors (Figure 7B). Taken together, these findings demonstrate that activation of the autophagy-mediated cell death pathway is indispensable for cannabinoid antitumoral action. Autophagy is essential for cannabinoid antitumoral action. (A) Effect of peritumoral THC administration on the growth of Atg5+/+ (upper panel) and Atg5–/– (lower panel) RasV12/T-large antigen MEF tumor xenografts generated in nude mice (mean ± SD; n = 7 for each condition). Photographs show representative images of vehicle- and THC-treated tumors. (B) Left: Effect of THC administration on LC3 immunostaining (green) and apoptosis as determined by TUNEL (red) in Atg5+/+ and Atg5–/– MEF tumor xenografts. Representative images from 1 vehicle-treated and 1 THC-treated Atg5+/+ and Atg5–/– tumors are shown. Right: Bar graphs show the percentage of TUNEL-positive nuclei and cells with TUNEL-positive nuclei and LC3 dots relative to the total number of nuclei in each section (mean ± SD). Eighteen sections were counted from 3 dissected tumors for each condition (vehicle-treated and THC-treated). Scale bar: 50 μm. (C) Schematic of the proposed mechanism of THC-induced cell death (see text for details). **P < 0.01 compared with vehicle-treated tumors. Finally, we analyzed the tumors of 2 patients enrolled in a clinical trial aimed at investigating the effect of THC on recurrent glioblastoma multiforme. The patients were subjected to intracranial THC administration, and biopsies were taken before and after the treatment (11). In the 2 patients, cannabinoid inoculation increased TRB3 immunostaining and decreased S6 phosphorylation (Figure 8A). Interestingly, the number of cells with autophagic phenotype (Figure 8B) as well as with active caspase-3 immunostaining (Figure 8C) was increased in the tumor samples obtained after THC treatment. Although these studies were only conducted in specimens from 2 patients, they are in line with the preclinical evidence shown above and suggest that cannabinoid administration might also trigger autophagy-mediated cell death in human tumors. THC administration promotes autophagy in glioblastomas of 2 patients. Analysis of different parameters in 2 patients with glioblastoma multiforme before and after intracranial THC treatment (it was estimated that doses of 6–10 μM were reached at the site of administration). (A) TRB3 and p-S6 immunostaining. Representative photomicrographs are shown. Numbers indicate the TRB3- or p-S6–stained area normalized to the total number of nuclei in each section (mean fold change ± SD) relative to the corresponding pre-treatment sample. Fifteen sections were counted for each tumor and each condition (before and after treatment). Scale bar: 50 μm. (B) Representative photomicrographs of LC3 diaminobenzidine immunostaining. The mean percentage of cells with LC3 dots ± SD relative to the total number of nuclei in each section is noted in the corner of each panel. Ten sections were counted from each biopsy for each condition. Arrows point to cells with LC3 dots. Scale bar: 20 μm. (C) Representative photomicrographs of active caspase-3 diaminobenzidine immunostaining. Numbers indicate the percentage of cells with active caspase-3 staining ± SD relative to the total number of nuclei in each section. Ten sections were counted from each biopsy for each condition. Arrows point to cells with active caspase-3 staining. Scale bar: 20 μm. *P < 0.05 and **P < 0.01 compared with before treatment. DiscussionIn this study we show that cannabinoids, a new family of potential antitumoral agents, induce autophagy of cancer cells and that this process mediates the cell death–promoting activity of these compounds. Several observations strongly support this idea: (a) THC induced autophagy and cell death in different types of cancer cells but not in nontransformed astrocytes, which are resistant to cannabinoid killing action, (b) pharmacological or genetic inhibition of autophagy prevented THC-induced cell death, (c) autophagy-deficient tumors were resistant to THC growth-inhibiting action, and (d) THC administration activated the autophagic cell death pathway in 3 different models of tumor xenografts as well as in 2 human tumor samples. Depending on the cellular context and the strength and duration of the triggering stimulus, autophagy is involved in the promotion or inhibition of cancer cell survival (4, 5, 24, 25). However, the molecular bases of this dual role of autophagy in cancer remain unknown. Data presented here demonstrate that induction of autophagy by cannabinoids leads to cancer cell death and identify the signaling route responsible for the activation of this cellular process. Thus, our findings suggest that THC — via activation of the CB1 receptor and stimulation of ceramide synthesis de novo — activates an early ER stress response that leads to increased phosphorylation of eIF2α on Ser51. Experiments performed with eIF2α S51A mutant cells have shown that phosphorylation of this residue, which is known to attenuate general protein translation while enhancing the expression of several genes related with the ER stress response (17), is required for the upregulation of the stress protein p8 and its ER stress–related downstream targets ATF4, CHOP, and TRB3 as well as for the induction of autophagy by cannabinoids. Furthermore, we demonstrate that the upregulation of p8 and TRB3, which has been previously implicated in cannabinoid-evoked cell death (13), is a crucial event in the triggering of autophagy. Ceramide accumulation has been proposed to induce ER stress (26, 27) and autophagy (28), and eIF2α phosphorylation has been implicated in the induction of autophagy in response to different situations (29–31). However, the molecular mechanisms responsible for these actions have not been clarified. Findings presented here now suggest that upregulation of the p8-TRB3 pathway constitutes a mechanism by which de novo–synthesized ceramide and eIF2α phosphorylation promote autophagy, thus identifying what we believe is a novel connection between ER stress and autophagy. Our data also demonstrate that the autophagy-promoting activity of the p8-regulated pathway is based on its ability to inhibit the Akt/mTORC1 axis. Regulation of mTORC1 largely relies on the activity of the prosurvival kinase Akt, whose inhibition leads to mTORC1 inactivation and, in turn, to autophagy (20). Our findings reveal that THC upregulates TRB3, promoting its interaction with Akt and leading to decreased phosphorylation of this kinase as well as of its direct substrates TSC2 and PRAS40, which triggers mTORC1 inhibition and induction of autophagy. TRB3 has been previously shown to inhibit Akt (21, 22), although the precise contribution of this pseudo-kinase to the regulation of Akt activity in different cellular contexts is unclear (32). Here we demonstrate that TRB3 inhibition of the Akt/mTORC1 axis is essential for cannabinoid-induced autophagy of cancer cells. Moreover, we show that this pathway is essential for cannabinoid antitumoral action. Thus, THC administration leads to TRB3 upregulation, mTORC1 inhibition, induction of autophagy, and reduction of tumor growth in different models of tumor xenografts, but not in p8-deficient tumors that are defective in the upregulation of the p8/TRB3 pathway. Furthermore, activation of this pathway was also evident in 2 glioma patients that had been treated with THC. These results thus uncover a role for TRB3 that may be of great importance in the regulation of cancer cell death. Autophagy has been proposed to protect from apoptosis, act as an apoptosis-alternative pathway to induce cell death, or act together with apoptosis as a combined mechanism for cell death (6, 33). However, very little is known about the role of the interplay between these 2 cellular processes in the control of tumor growth in response to anticancer agents. Our results now clearly demonstrate that induction of autophagy is involved in the mechanism by which cannabinoids promote the activation of the mitochondrial pro-apoptotic pathway. Thus, neither tumors in which the p8-regulated pathway has been ablated (and in which, therefore, THC treatment does not induce autophagy) nor tumors intrinsically deficient in autophagy undergo apoptosis in response to THC, and so they are resistant to THC antitumoral action. These findings reveal that autophagy is required for the activation of apoptosis in response to cannabinoid treatment in vivo. It is worth noting that the concentrations of THC used in this study are in the same range as those administered intracranially to the patients in which we observed activation of the autophagy-mediated cell death pathway (11) and could be thus considered clinically relevant. Of interest, intraperitoneal administration of THC to U87MG tumor xenografts produces a similar decrease in tumor growth (that occurs in concert with increased autophagy and apoptosis) to that observed when the cannabinoid is administered peritumorally (our unpublished observations). Considering that no signs of toxicity were observed in the clinical trial patients (11) or in tumor-bearing animals treated intracranially, peritumorally, or intraperitoneally with THC (refs. 34 and 35 and data not shown), and that no overt toxic effects have been reported in other clinical trials of cannabinoid use in cancer patients for various applications (e.g., inhibition of nausea, vomiting, and pain) and using different routes of administration (e.g., oral, oro-mucosal) (9, 36), our findings support that safe, therapeutically efficacious doses of THC may be reached in cancer patients. In summary, in this study we identify what we believe is a new route that links the ER stress response to the activation of autophagy and promotes the apoptotic death of tumor cells (Figure 7C). The identification of this pathway will help to understand the molecular events that lead to activation of autophagy-mediated cell death by anticancer drugs and may contribute to the design of new therapeutic strategies for inhibiting tumor growth. MethodsCell culture and viability. Cortical astrocytes were prepared from 24-hour-old mice as previously described (13). Primary cultures of brain tumor cells were prepared and cultured as described in the Supplemental Methods. U87MG, T98G, U373MG, and MiaPaCa2 cells, p8+/+ and p8–/– RasV12/E1A MEFs, Atg5+/+ and Atg5–/– T-large antigen MEFs (provided by Noboru Mizushima, Tokyo Medical and Dental University, Tokyo, Japan), Bax/Bak wild-type and Bax/Bak DKO T-large antigen MEFs (provided by Luca Scorrano, Dulbecco Telethon Institute, Milan, Italy, and Patrizia Agostinis, Catholic University of Leuven, Leuven, Belgium), eIF2α S51S WT and eIF2α S51A T-large antigen MEFs (provided by Richard Kaufman, University of Michigan, Ann Arbor, Michigan, USA, and Cesar de Haro and Juan J. Berlanga, Centro de Biología Molecular Severo Ochoa, Autonoma University, Madrid, Spain), Tsc2+/+ and Tsc2–/– p53–/– MEFs, empty vector (pBABE) and pBABE-myr-Akt MEFs, and Atg5+/+ and Atg5–/– RasV12/T-large antigen MEFs were cultured in DMEM containing 10% FBS and transferred to medium containing 0.5% FBS (except RasV12/E1A-transformed MEFs, which were transferred to medium containing 2% FBS) 18 h before performing the different treatments. p8+/+ and p8–/– RasV12/E1A MEFs as well as Atg5+/+ and Atg5–/– RasV12/T-large antigen MEFs correspond to a polyclonal mix of at least 20 different selected clones. Unless otherwise indicated, THC was used at a final concentration of 5 μM. Cell viability was determined by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] test (Sigma-Aldrich). Flow cytometry. Briefly, cells (approximately 5 × 105 cells per assay) were trypsinized, divided in 2 tubes, washed, and collected by centrifugation at 1,500 g for 5 min. One aliquot was incubated for 10 min at 37°C with Annexin V–FITC (BD Biosciences). Propidium iodide (1 μg/ml) was added just before cytofluorometric analysis. The other aliquot was simultaneously labeled with 3,3ι-dihexyloxacarbocyanine iodide (DiOC6[3], 40 nM; Invitrogen) and hydroethidium (5 μM; Invitrogen) for 10 minutes at 37°C, followed by cytofluorometric analysis. Cells (10,000) were recorded in each analysis. Fluorescence intensity was analyzed in an EPICS XL flow cytometer (Beckman Coulter). Western blot. Western blot analysis was performed following standard procedures. A list of the antibodies used can be found in Supplemental Methods. Densitometric analysis was performed with Quantity One software (Bio-Rad). Transfections. U87MG cells (75% confluent) were transfected with siRNA duplexes using the DharmaFECT 1 Transfection reagent (Dharmacon). Cells were trypsinized and seeded 24 h after transfection, at a density of 5,000 cells/cm2. Transfection efficiency was greater than 70% as monitored with a control fluorescent (red) siRNA (siGLO RISC-Free siRNA; Dharmacon). In immunofluorescence experiments, control and selective siRNAs were used in a 1:5 ratio, and cells with red spots were scored as transfected. Infections with adenoviral vectors. U87MG cells (75% confluent) were transduced for 1 h with supernatants obtained from HEK293 cells infected with adenoviral vectors carrying EGFP (provided by Javier G. Castro, Hospital Infantil Universitario Niño Jesús, Madrid, Spain), rat HA-tagged TRB3 (donated by Patrick Iynedjian, University of Geneva, Geneva, Switzerland) (32), or human EGFP-LC3 (provided by Aviva Tolkovsky and Christoph Goemans, University of Cambridge, Cambridge, United Kingdom). Infection efficiency was greater than 80% as determined by EGFP fluorescence. RNA interference. Double-stranded RNA duplexes were purchased from Dharmacon. A list of sequences can be found in the Supplemental Methods. RT-PCR analysis. RNA was isolated using Trizol Reagent (Invitrogen). cDNA was obtained with Transcriptor Reverse transcriptase (Roche Applied Science). Primers and amplification conditions can be found in the Supplemental Methods. Real-time quantitative PCR. cDNA was obtained using Transcriptor (Roche Applied Science). Real-time quantitative PCR assays were performed using the FastStart Universal Probe Master mix with Rox (Roche Applied Science), and probes were obtained from the Universal ProbeLibrary Set (Roche Applied Science). Primer sequences can be found in the Supplemental Methods. Amplifications were run in a 7900 HT-Fast Real-Time PCR System (Applied Biosystems). Each value was adjusted by using 18S RNA levels as a reference. Immunoprecipitation. U87MG cells were lysed in HEPES lysis buffer (see Supplemental Methods for buffer composition). Lysate (1–4 mg) was precleared by incubating with 5–20 μl of protein G–Sepharose conjugated to pre-immune IgG. The lysate extracts were then incubated with 5–20 μl of protein G–Sepharose conjugated to 5–20 μg of the anti-TRB3 antibody or pre-immune IgG. TRB3 antibody (aminoterminal end, ab50516; Abcam) was covalently conjugated to protein G–Sepharose using dimethyl pimelimidate. Immunoprecipitations were carried out for 1 h at 4°C on a rotatory wheel. The immunoprecipitates were washed 4 times with HEPES lysis buffer, followed by 2 washes with HEPES kinase buffer. The immunoprecipitates were resuspended in 30 μl of sample buffer (not containing 2-mercaptoethanol) and filtered through a 0.22-μm Spin-X filter, and 2-mercaptoethanol was added to a concentration of 1% (vol/vol). Samples were subjected to electrophoresis and immunoblot analysis. Ceramide levels. Ceramide levels were determined as previously described (37). Confocal laser scanning microscopy. Standard protocols for immunofluorescence microscopy were used (see Supplemental Methods for the antibodies used). To quantify the percentage of cells with LC3 or PDI dots, at least 200 cells per condition were counted in randomly selected fields. In all cases, only those cells with 4 or more prominent dots of either LC3 or PDI were scored positively. In vivo treatments. Tumors derived from U87MG cells and p8+/+ and p8–/– MEFs were induced and treated as previously described (13). Tumors derived from Atg5+/+ or Atg5–/– RasV12/T-large antigen MEFs (see Supplemental Methods for the procedure used to generate these cells) were induced in nude mice by subcutaneous injection of 107 cells in PBS supplemented with 0.1% glucose. Tumors were allowed to grow until an average volume of 200–250 mm3, and animals were assigned randomly to the different groups. At this point, vehicle or THC (15 mg/kg/d) in 100 μl of PBS supplemented with 5 mg/ml BSA was administered daily in a single peritumoral injection. Tumors were measured with an external caliper, and volume was calculated as (4π/3) × (width/2)2 × (length/2). All procedures involving animals were performed with the approval of the Complutense University Animal Experimentation Committee according to Spanish official regulations. Human tumor samples. Tumor biopsies were obtained from 2 recurrent glioblastoma multiforme patients who had been treated with THC. The characteristics of the patients and the clinical study have been described in detail elsewhere (11). Briefly, THC dissolved in 30 ml of physiological saline solution plus 0.5% (wt/vol) human serum albumin was administered intratumorally to the patients. Patient 1 received a total of 1.46 mg of THC for 30 days, while patient 2 received a total of 1.29 mg of THC for 26 days (it was estimated that doses of 6–10 μM THC were reached at the site of administration; ref. 11). Samples were fixed in formalin, embedded in paraffin, and used for immunomicroscopy. Immunomicroscopy of tumor samples. Samples from tumor xenografts were dissected, Tissue-Tek (Sakura) embedded, frozen, and, before the staining procedures were performed, fixed in acetone for 10 min at room temperature. Samples from human tumors were subjected to deparaffinization, rehydration, and antigen retrieval before the staining procedures were performed. Standard protocols for immunofluorescence or immunohistochemistry microscopy were used (see Supplemental Methods). Nuclei were counterstained with TOTO-3 iodide (U87MG and human tumor samples; Invitrogen) or Hoechst 33342 (MEF tumors; Invitrogen). Fluorescence images were acquired using Metamorph-Offline 6.2 software (Universal Imaging) and Zeiss Axioplan 2 Microscope. TUNEL. Tumor samples were fixed, blocked, and permeabilized, and TUNEL was performed as previously described (13). Electron microscopy. Ultrastructural analysis of vehicle- and THC-treated cells was assessed by conventional embedding in the epoxy-resin EML-812 (Taab Laboratories). Ultrathin (20- to 30-nm-thick) sections of the samples were obtained using a Leica-Reichert-Jung ultramicrotome and then stained with saturated uranyl acetate–lead citrate by standard procedures. Ultrathin sections were analyzed in a JEOL 1200-EX II transmission electron microscope operating at 100 kV. Statistics. Statistical analysis was performed by ANOVA with a post-hoc analysis using the Student-Neuman-Keuls test. Differences were considered significant when the P value was less than 0.05. Supplemental dataAcknowledgmentsThis work was supported by grants from the Spanish Ministry of Education and Science (MEC) (HF2005/0021, to G. Velasco; SAF2006/00918, to M. Guzmán; and BFU2006-00508, to P. Boya), Santander-Complutense PR34/07-15856, to G. Velasco), Comunidad de Madrid (S-SAL/0261/2006, to M. Guzmán), and La Ligue contre le Cancer and Canceropole PACA (to J.L. Iovanna). M. Salazar was the recipient of a fellowship from the MEC. A. Carracedo was the recipient of fellowships from Gobierno Vasco, the Federation of European Biochemical Societies, and the European Molecular Biology Organization. M. Lorente and P. Boya have a Juan de la Cierva and a Ramón y Cajal contract from the MEC, respectively. S. Hernández-Tiedra has a technician contract from the Spanish Ministry of Education and the Fondo Social Europeo. The authors thank Dario Alessi (University of Dundee, Dundee, United Kingdom) for donating anti-PRAS40 antibodies and for technical support for immunoprecipitation experiments; Gemma Fabriàs, Josefina Casas, and Eva Dalmau (Instituto de Investigaciones Químicas y Ambientales, Barcelona, Spain) for analyzing ceramide samples; José Lizcano, José Bayascas, María M. Caffarel, and Patrizia Agostinis for their experimental suggestions; and other members of our laboratory for their continual support. FootnotesConflict of interest: The authors have declared that no conflict of interest exists. Nonstandard abbreviations used: Atg, autophagy protein; eIF2α, eukaryotic translation initiation factor 2α; MEF, mouse embryonic fibroblast; THC, Δ9-tetrahydrocannabinol; mTORC1, mammalian target of rapamycin complex 1; PDI, protein disulphide isomerase; TRB3, tribbles homolog 3. Citation for this article: J. Clin. Invest. 119:1359–1372 (2009). doi:10.1172/JCI37948 Arkaitz Carracedo and Ainara Egia’s present address is: Cancer Genetics Program, Beth Israel Deaconess Cancer Center and Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA. ReferencesView this article via: CrossRef
View this article via: PubMed
View this article via: PubMed
View this article via: PubMed
View this article via: PubMed
View this article via: PubMed
( categories:
|
Support Civic Journalism Recent commentsPopular contentToday's:
All time:Last viewed:
|




Live in Ohio and get cancer... or
Live in Ohio and get cancer... or live in any of 16 other states or DC and get rid of cancer. Not a hard decision.
Disrupt IT
Considering that no signs of toxicity were observed
Considering that no signs of toxicity were observed in the clinical trial patients (11) or in tumor-bearing animals treated intracranially, peritumorally, or intraperitoneally with THC (refs. 34 and 35 and data not shown), and that no overt toxic effects have been reported in other clinical trials of cannabinoid use in cancer patients for various applications (e.g., inhibition of nausea, vomiting, and pain) and using different routes of administration (e.g., oral, oro-mucosal) (9, 36), our findings support that safe, therapeutically efficacious doses of THC may be reached in cancer patients.
Name a DRUG that does ANY of THAT.
Disrupt IT
Please don't let any of this research happen in Cleveland
Please don't let any of this research happen in Cleveland, Mr. President... we don't need good jobs here, and this may effect the bad jobs of people holding the hands of those dying of cancer here... and the hospitals soaking the welfare state... and the government shills managing the medicaid for all that.
Disrupt IT